Materials Science | ; Simulation, Learning
Towards DMC Accuracy Across Chemical Space with Scalable Δ-QML
Improving our ability to understand and predict the behavior and properties of molecules and materials is crucial to enabling the design and discovery of new materials for batteries, catalysts, semiconductors, and countless other applications. With this INCITE project, a multi- institutional team is advancing the use of quantum Monte Carlo (QMC) methods, coupled with machine learning, to provide accurate and reliable predictions of the fundamental properties of a wide range of molecules and materials.
Challenge
The predictive accuracy of quantum machine learning (QML) models trained on quantum chemistry data and used for the navigation of the chemical compound space is inherently limited by the predictive accuracy of the approximations used within the underlying quantum theory. To help QML models achieve the coveted threshold of chemical accuracy (~1 kcal/mol average deviation of calculated values from experimental measurements of atomization energies), the INCITE team is leveraging DOE supercomputers to demonstrate the usefulness of recently implemented and numerically efficient QMC methods for generating highly accurate training data.
Approach
The team’s primary application is QMCPACK, an open-source code for computing the electronic structure of atoms, molecules, 2D nanomaterials, and solids. As part of a recent study, the researchers used the ALCF’s Theta supercomputer to couple QMCPACK with Δ-QML-based surrogate methods to predict the energetics of large molecules at chemical accuracy and at a fraction of the computational cost of traditional machine learning methods.
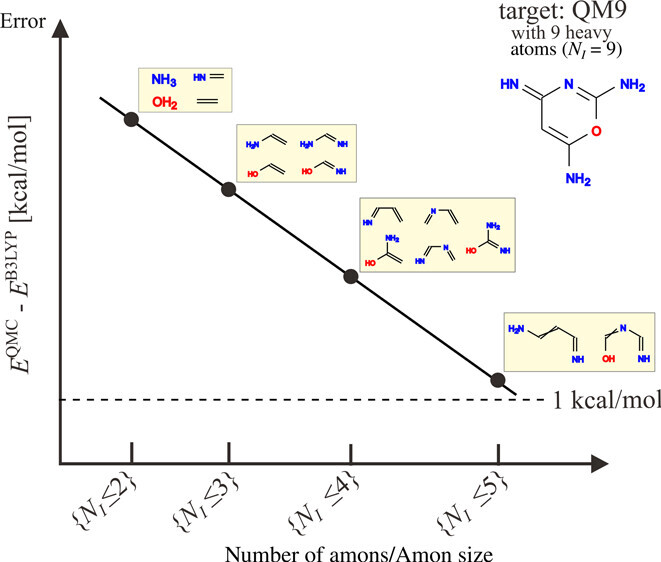
Results
In a paper published in the Journal of Chemical Theory and Computation, the team showed that their Δ-QML framework can alleviate the computational burden of QMC such that it offers clear potential to support the formation of high-quality descriptions across the chemical space. Their work involved using Theta to conduct diffusion Monte Carlo (DMC) calculations on over 1,000 small amons containing up to five heavy atoms and covering parts of the QM9 database, which is used routinely for machine learning predictions of various chemical properties. This is the largest dataset ever computed with DMC and the first use of such a dataset for machine learning. The team’s research suggests that the QMC training datasets of amons can predict total energies with near chemical accuracy throughout chemical space, setting the foundation for the study of larger databases.
Impact
Using the Δ-QML approach, the team was able to predict the energetics of large molecules at a reduced computational cost while maintaining chemical accuracy. The high efficiency of the Δ-QML framework compared to traditional approaches indicates a path to use the computationally expensive but highly accurate QMC methodology in machine learning. This new method will allow researchers to study larger systems and predict the properties of molecules and materials more accurately, which could lead to significant advances in fields such as materials science, drug discovery, and energy research.